




Progress for Medicine, Chemistry, and Materials Science
Researchers from Google DeepMind in Berlin, BIFOLD, and the Technical University of Berlin have introduced a new machine learning method that enables global atomic interactions in chemical systems to be represented more efficiently. This could allow chemical and materials science processes to be simulated more accurately in the future, potentially accelerating the development of new drugs, more efficient batteries, and more sustainable materials. The work, titled “Machine Learning Global Atomic Representations with Euclidean Fast Attention,” was published in Nature Machine Intelligence in March 2026.
To understand exactly how, for example, a drug works, scientists must precisely calculate how atoms in molecules move and interact with one another. Such simulations form the foundation of modern drug development, as well as the design of new materials and more efficient catalysts. However, many computational methods reach their limits when dealing with larger molecules containing hundreds or thousands of atoms due to their complexity. Modeling atomistic systems is challenging because each atom simultaneously experiences forces from many other atoms, including some that are far away, not just from its immediate neighbors. This results in a highly complex many-body system in which even small changes in one location can affect the behavior of the entire system.
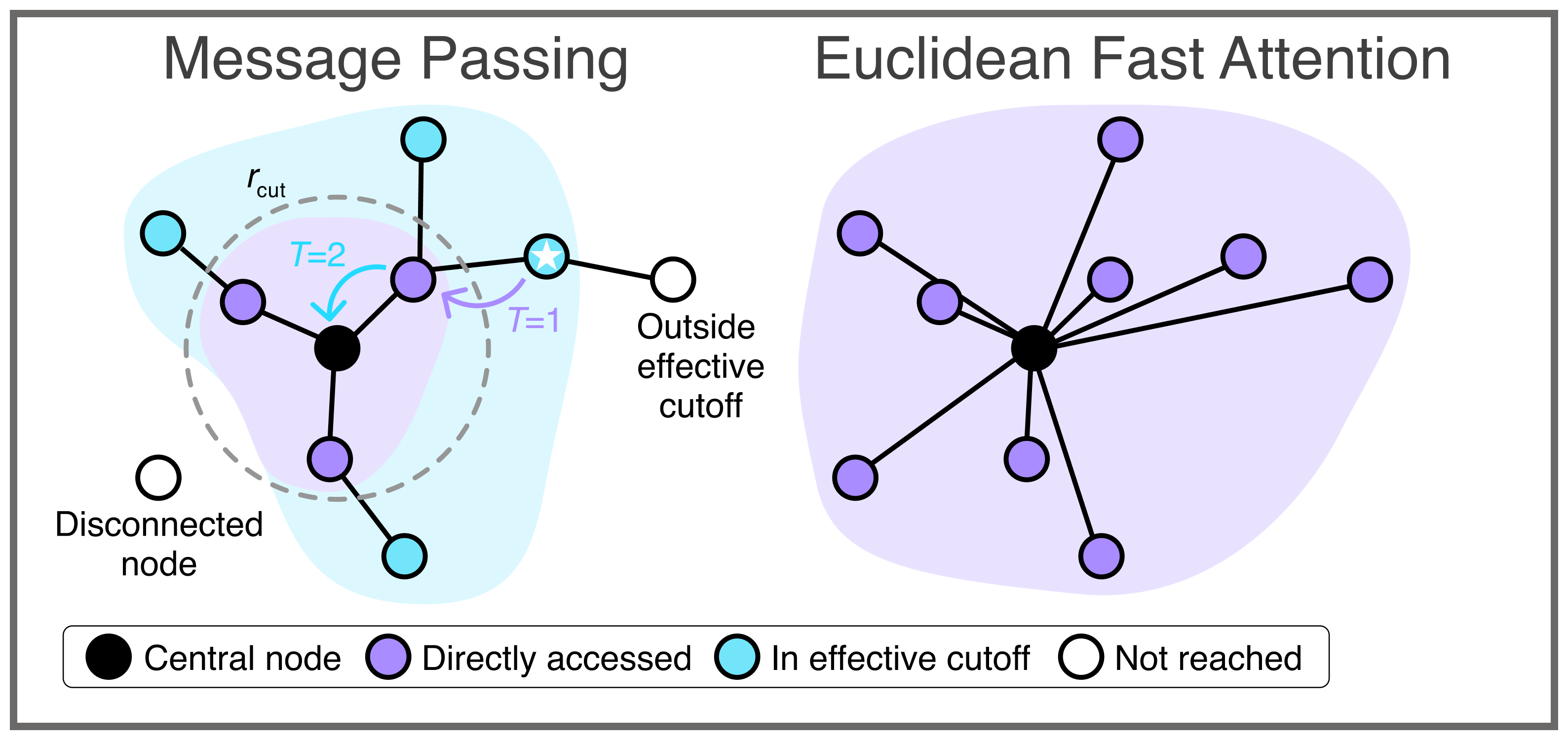
The new representation of these interactions is called Euclidean Fast Attention (EFA)
A central role in this process is played by a fundamental concept in modern machine learning known as self-attention. This concept enables models to assess the importance of individual pieces of information in the context of all other information, thereby capturing long-range relationships. However, as the number of atoms increases, the number of relevant interactions grows approximately with the square of the number of atoms. This makes the use of self-attention for precise modeling of physical systems extremely computationally expensive and limits the size of atomistic structures that can be simulated at all.
This is exactly where the research team’s new method comes into play. The scientists developed a new, linearly scaling representation of these interactions called Euclidean Fast Attention (EFA). It was specifically designed for data in Euclidean space, where the rules of classical geometry apply, for example, atoms in molecules and materials, whose relative positions and orientations are crucial for accurate predictions. A key aspect of the approach is that spatial information can be represented efficiently without violating important physical symmetries. In their experiments, the researchers show that EFA effectively captures different long-range effects and can describe chemical interactions for which conventional machine-learning force fields may produce incorrect results. This makes it possible to reliably capture interactions over large distances, while requiring comparatively low computational effort.
“Our approach enables an important new step toward more quantum-mechanically accurate modeling of many-body systems using new deep learning methods,” says Prof. Klaus-Robert Müller, co-director of BIFOLD and professor at the Technical University of Berlin.
The work therefore addresses a key question in modeling many-body systems in chemistry and physics: How can global structural information be incorporated into atomistic models without sacrificing the computational efficiency required for large systems? Because the method is specifically designed to work efficiently with large molecules, it can also be applied in the future to particularly demanding systems, such as large or complex materials. The authors view EFA as a promising approach for making machine learning methods more robust and more efficient for challenging chemical and materials science simulations.
Publication
Frank, J. T., Chmiela, S., Müller, K.-R., Unke, O., Machine Learning Global Atomic Representations With Euclidean Fast Attention. Nature Machine Intelligence 8, 388–402 (2026).
DOI: 10.1038/s42256-026-01195-y.