



The use of AI in classical sciences such as chemistry, physics, or mathematics remains largely uncharted territory. Researchers from the Berlin Institute for the Foundation of Learning and Data (BIFOLD) at TU Berlin and Google Research have successfully developed an algorithm to precisely and efficiently predict the potential energy state of individual molecules using quantum mechanical data. Their findings, which offer entirely new opportunities for material scientists, have now been published in the paper “SpookyNet: Learning Force Fields with Electronic Degrees of Freedom and Nonlocal Effects” in Nature Communications.
“Quantum mechanics, among other things, examines the chemical and physical properties of a molecule based on the spatial arrangement of its atoms. Chemical reactions occur based on how several molecules interact with each other and are a multidimensional process,” explains BIFOLD Co-Director Prof. Dr. Klaus-Robert Müller. Being able to predict and model the individual steps of a chemical reaction at the molecular or even atomic level is a long-held dream of many material scientists.
Every individual atom in focus
The potential energy surface, which refers to the dependence of a molecule’s energy on the arrangement of its atomic nuclei, plays a key role in chemical reactivity. Knowledge of the exact potential energy surface of a molecule allows researchers to simulate the movement of individual atoms, such as during a chemical reaction. As a result, they gain a better understanding of the atoms’ dynamic, quantum mechanical properties and can precisely predict reaction processes and outcomes. “Imagine the potential energy surface as a landscape with mountains and valleys. Like a marble rolling over a miniature version of this landscape, the movement of atoms is determined by the peaks and valleys of the potential energy surface: this is called molecular dynamics,” explains Dr. Oliver Unke, researcher at Google Research in Berlin.
Unlike many other fields of application of machine learning, where there is a nearly limitless supply of data for AI, generally only very few quantum mechanical reference data are available to predict potential energy surfaces, data which are only obtained through tremendous computing power. “On the one hand, exact mathematical modelling of molecular dynamic properties can save the need for expensive and time-consuming lab experiments. On the other hand, however, it requires disproportionately high computing power. We hope that our novel deep learning algorithm – a so-called transformer model which takes a molecule’s charge and spin into consideration – will lead to new findings in chemistry, biology, and material science while requiring significantly less computing power,” says Klaus-Robert Müller.
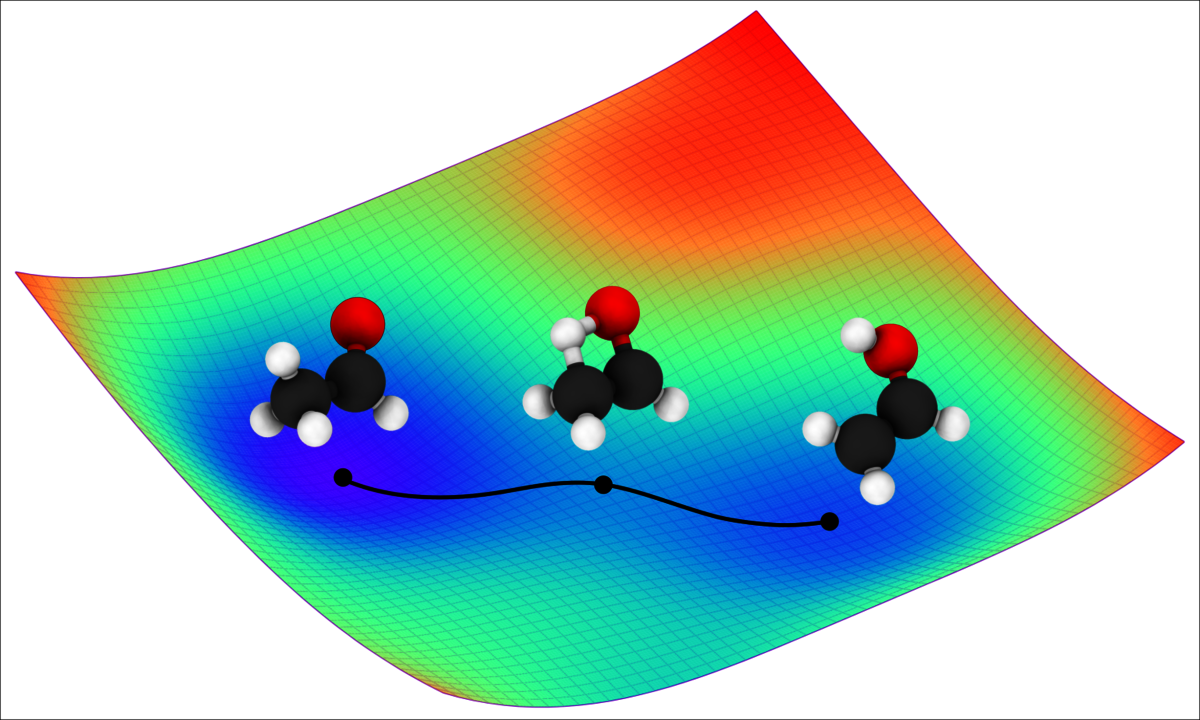
In order to achieve particularly high data efficiency, the researchers’ new deep learning model combines AI with known laws of physics. This allows certain aspects of the potential energy surface to be precisely described with simple physical formulas. Consequently, the new method learns only those parts of the potential energy surface for which no simple mathematical description is available, saving computing power. “This is extremely practical. AI only needs to learn what we ourselves do not yet know from physics,” explains Müller.
Spatial separation of cause and effect
Another special feature is that the algorithm can also describe nonlocal interactions. “Nonlocality” in this context means that a change to one atom, at a particular geometric position of the molecule, can affect atoms at a spatially separated geometric molecular position. Due to the spatial separation of cause and effect – something Albert Einstein referred to as “spooky action at a distance” – such properties of quantum systems are particularly hard for AI to learn. The researchers solved this issue using a transformer, a method originally developed for machine processing of language and texts or images. “The meaning of a word or sentence in a text frequently depends on the context. Relevant context-information may be located in a completely different section of the text. In a sense, language is also nonlocal,” explains Müller. With the help of such a transformer, the scientists can also differentiate between different electronic states of a molecule such as spin and charge. “This is relevant, for example, for physical processes in solar cells, in which a molecule absorbs light and is thereby placed in a different electronic state,” explains Oliver Unke.
The publication in detail:
Oliver T. Unke, Stefan Chmiela, Michael Gastegger, Kristof T. Schütt, Huziel E. Sauceda, Klaus-Robert Müller: SpookyNet: Learning force fields with electronic degrees of freedom and nonlocal effects. Nat. Commun. 12(7273) (2021)
Machine-learned force fields combine the accuracy of ab initio methods with the efficiency of conventional force fields. However, current machine-learned force fields typically ignore electronic degrees of freedom, such as the total charge or spin state, and assume chemical locality, which is problematic when molecules have inconsistent electronic states, or when nonlocal effects play a significant role. This work introduces SpookyNet, a deep neural network for constructing machine-learned force fields with explicit treatment of electronic degrees of freedom and nonlocality, modeled via self-attention in a transformer architecture. Chemically meaningful inductive biases and analytical corrections built into the network architecture allow it to properly model physical limits. SpookyNet improves upon the current state-of-the-art (or achieves similar performance) on popular quantum chemistry data sets. Notably, it is able to generalize across chemical and conformational space and can leverage the learned chemical insights, e.g. by predicting unknown spin states, thus helping to close a further important remaining gap for today’s machine learning models in quantum chemistry.
Machine-learned force fields combine the accuracy of ab initio methods with the efficiency of conventional force fields. However, current machine-learned force fields typically ignore electronic degrees of freedom, such as the total charge or spin state, and assume chemical locality, which is problematic when molecules have inconsistent electronic states, or when nonlocal effects play a significant role. This work introduces SpookyNet, a deep neural network for constructing machine-learned force fields with explicit treatment of electronic degrees of freedom and nonlocality, modeled via self-attention in a transformer architecture. Chemically meaningful inductive biases and analytical corrections built into the network architecture allow it to properly model physical limits. SpookyNet improves upon the current state-of-the-art (or achieves similar performance) on popular quantum chemistry data sets. Notably, it is able to generalize across chemical and conformational space and can leverage the learned chemical insights, e.g. by predicting unknown spin states, thus helping to close a further important remaining gap for today’s machine learning models in quantum chemistry.